药代动力学
性质
给药方便:口服有效,一次或两次/日(消炎镇痛药、抗高血压药物、抗菌药常用药)
靶向分布或靶向活化:抗肿瘤药物
起效快:抗过敏药物、镇痛药物
药物相互作用少:有利于联合用药,如降脂药与抗高血压药物的合用
长期使用不产生耐药性:如抗菌药、抗癌药、抗病毒药。
无蓄积:如果药物或其代谢物不能通过有效途径排出体外,会在体内蓄积,产生毒性.
重要性
随着药物化学的发展及人类健康水平的不断提高,对药物的药代动力学性质的要求越来越高:判断一个药物的应用前景特别是市场前景,不单纯是疗效强,毒副作用小;更要具备良好的药代动力学性质。肽类药物就是最典型的例子。一般来说,体内的许多生物活性肽如内啡肽等均具有高效低毒的特点,但是,体内不稳定,口服无效。
理化性质
代谢过程 吸收:药物口服后,进入消化道,在不同部位,如口腔、胃、肠吸收,进入血液。
分布:进入血液的药物进入作用部位,产生治疗作用或毒副作用。
代谢转化:药物在肝脏或胃肠道通过酶催化的一系列氧化还原反应发生生物转化。
排泄:药物或代谢物经肾(尿)或胆汁(粪)或呼吸排泄。
为了表述的方便,常把体内过程分为三个时相:
药剂相:片剂或胶囊崩解、溶出,成为可被吸收的形式。(药剂学研究内容。)
药代动力相:药物吸收、分布、代谢与排泄。(药代动力学研究内容。)
药效相:药物与作用靶点相互作用,通过刺激和放大,引发一系列的生物化学和生物物理变化,导致宏观上可以观察到的活性或毒性。(药理学或毒理学研究内容。)
三个时相依次发生,但是可能同时存在:如缓释药物,一部分药物已完成分布、发挥药理作用,但是另一部分还在释放和吸收的过程中。特别是药代动力相和药效相一般同时存在。
药代动力参数
一、吸收
溶出度:药物分子在消化道中溶解的程度
生物利用度:药物吸收的程度
绝对生物利用度
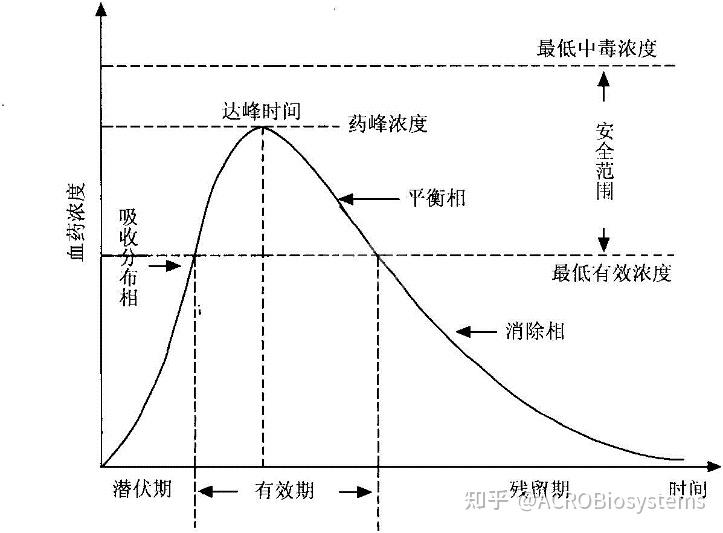
最大血药浓度(Cmax)
达峰时间(Tmax)
二、分布
由于体内环境的非均一性(血液、组织),导致药物浓度变化的速度不同。
隔室(compartment):同一隔室药物浓度的变化速度相同,均相。
一室模型:药物进入血液迅速分布全身,并不断被清除。
二室模型: 药物进入体内后,首先快速分布于组织中,然后进入较慢的消除过程。
表观分布体积(Vd)(aparent volume of distribution):表征药物在体内被组织摄取的能力。表观容积大的药物体内存留时间较长。
药物浓度-时间曲线下面积(AUC);系统药物暴露(Systemic Exposure)
血脑屏障;蛋白结合率;分布半衰期(t 1/2(α)
三、消除
消除(elimination):原药在体内消失的过程。包括肾(尿)或胆汁(粪)或呼吸排泄及代谢转化的总和。
消除速率常数(elimination constants): 反映药物在体内消失的快慢。不完全反映药物的作用时间(代谢物也有活性)。
半寿期或半衰期(t1/2):药物浓度或药量降低50%所需的时间。消除半衰期t1/2(β))Terminal Half-life ,Elimination Half-life。
清除率(clearance,廓清率)或肾清除率(renal clearance):反映药物或代谢物经肾被 排出体外的速度。
相互作用 一方面是药物对机体的作用,产生药效、毒性或副作用,表现为药物的药理作用或毒理作用,决定于特定的化学结构,具有较强的结构特异性。
另一方面是机体对药物的作用:吸收、分布,生物转化和排泄,表现为药物的药代动力学性质。主要取决于药物的溶解性、脂水分配系数、电荷等药物分子整体的理化性质,结构特异性不强。
吸收
药物的吸收 是药物由给药部位通过生物膜进入血液循环的过程。
吸收部位 消化道(口服给药,口腔、胃、小肠、大肠)、呼吸道(鼻腔给药,肺)、肌肉(肌肉注射)、粘膜(栓剂)。
吸收部位不同,药物被吸收的程度和快慢,有差异(静注、肌注;皮下给药,口服。)
共性: 药物是通过生物膜吸收的。
吸收过程 扩 散
被动扩散:扩散速率与浓度梯度成正比;无特异性;无饱和性;药物分子必须具备合适的脂水分配系数。大部分化学药物是通过被动扩散途径吸收的。 药物分布 亲脂性:向组织分布,必须通过细胞膜。
合适的脂水分配系数。
电荷: 带电分子难以通过细胞膜和血脑屏障与组织或蛋白的结合,与血浆蛋白结合,不能穿越细胞膜或血管壁,不能扩散入细胞内,也不能被肾小球过滤,影响分布容积、生物转化和排泄速度。
血浆蛋白结合 与血浆蛋白的结合能够维持较平稳的血药浓度,因此调整药物分子中的非活性必须结构,可以改变结合与解离的平衡,延长药物的作用时间。
由于这是一种非特异性结合的可逆性结合,不直接影响疗效,但是影响药代过程,因而间接地影响受体部位的药物有效浓度。
不能被肾小球过滤,影响分布容积、生物转化和排泄速度。
折叠影响因素 亲脂性强的药物与组织蛋白或脂肪组织的亲和力高,结合较强:起长效作用。
烷基、芳环基、卤素等疏水性基团,增加与蛋白的结合亲和力。
可离解性的药物,也可通过电荷相互作用与蛋白结合。
药物的立体结构影响药物与血浆蛋白的结合。
手性药物的不同光学异构体具有不同血浆蛋白结合作用。
特异性分布 利用某些组织对特定配基的选择性识别和结合作用,将药物分子与这些配基偶联,将药物分子选择性投放于特定的组织,以提高药物作用的选择性。
靶向给药
主动靶向:如抗体导向药物;受体靶向药物
被动靶向:利用组织的屏障作用,将药物包裹于脂质体或微球中,避免药物向非作用部位分布,以免代谢失活或产生毒副作用。
参考文献
- ↑ 药代动力学主要研究什么百度知道