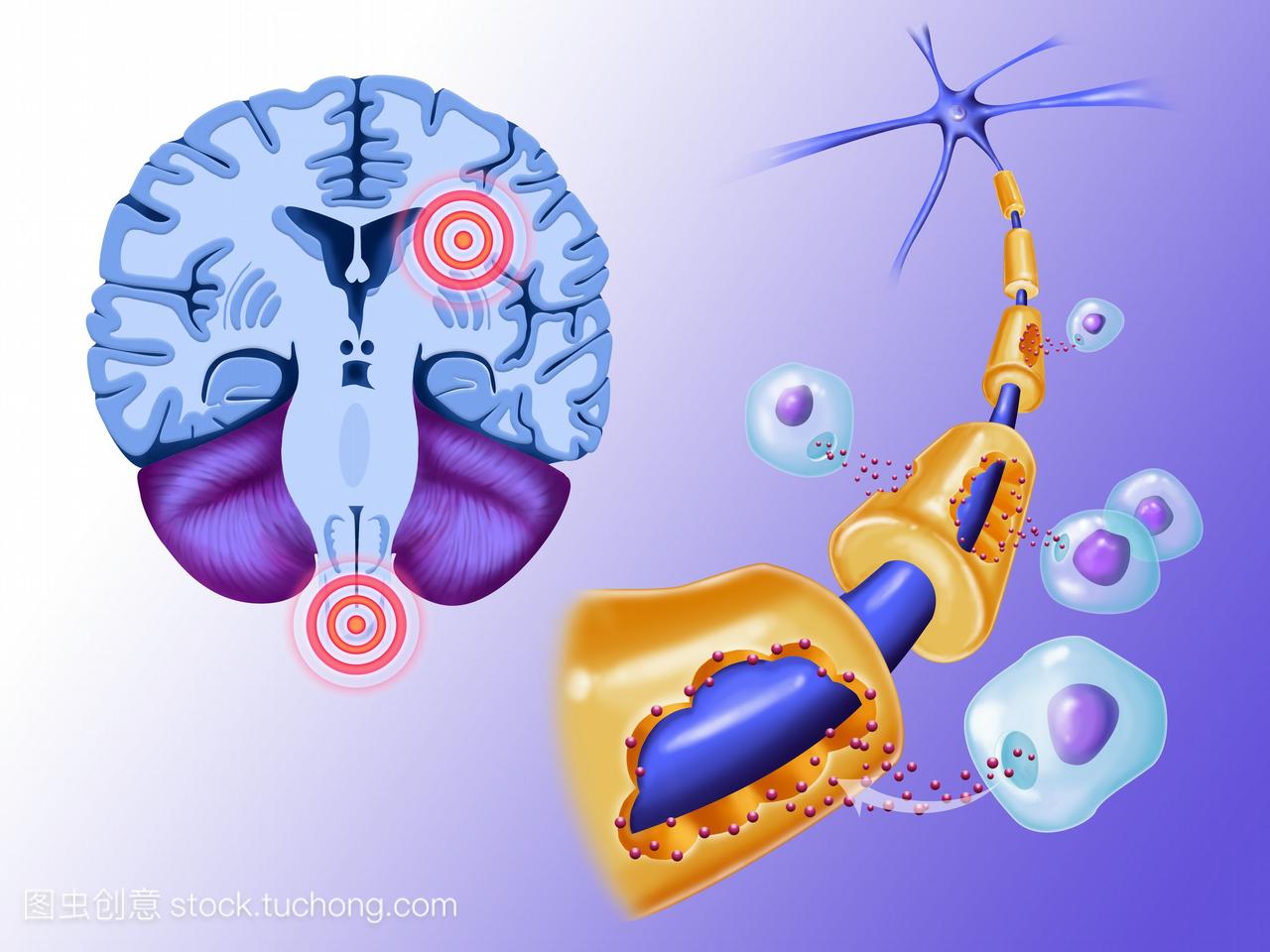
多發性硬化

{kind=link}

{kind=link}

{kind=link}
多發性硬化(multiple sclerosis,MS)是中樞神經系統脫髓鞘疾病中最常見最主要的疾病,以中樞神經系統(CNS)白質脫髓鞘病變為特點,遺傳易感個體與環境因素作用發生的自身免疫性疾病。患者以青、中年多見,臨床特點是病灶播散廣泛,病程中常有緩解復發的神經系統損害症狀。
多發性硬化作為獨立的疾病已有100餘年的歷史。因其有較高的發病率、慢性病程和青壯年易患而備受重視。其臨床特徵為發作性視神經、脊髓和腦部的局灶性障礙。這些神經障礙可有不同程度的緩解、復發。[1]
目錄
病因
確切的病因尚不肯定,但與遺傳因素,環境因素如病毒感染、地理位置、自身免疫反應有一定關係。
病理改變
腦外觀一般是正常的,但脊髓的表面可呈現不均勻。腦和脊髓切面顯現大量散在、輕度下陷、因髓鞘脫失而呈粉紅色的白質病灶。病灶大小從1mm到數厘米不等;這些病灶基本位於腦和脊髓的白質,不超過脊神經和腦神經根的進入區。因其具有明顯的輪廓,法國病理學家稱之為硬化斑。
病損的分布有一定的規律,病變多位於腦室周圍,特別是在腦室周圍的室管膜下靜脈走行處(主要靠近腦室體部和側腦室角部)。其他易受侵犯的結構為視神經和視交叉(但很少影響視束)、脊髓的軟膜靜脈與白質相鄰處。病損任意地分布於腦幹、脊髓和小腦臂而不偏重於某一種纖維。在腦皮質、中央核團和脊髓結構,病變破壞髓鞘但神經細胞則相對保留完好。
病灶組織學的表現形式取決於病變產生的時間。相對新的病灶由許多位於靜脈周圍的灶性脫髓鞘區融合而成,病灶區髓鞘部分或全部破壞或脫失,而軸突相對保留。有不同程度的少突膠質細胞變性,膠質細胞反應(星形細胞)和血管周圍、外膜旁單核細胞和淋巴細胞滲出。後期,大量的小膠質吞噬細胞(巨噬細胞)浸潤,病灶內以及周圍的星形細胞體積增大、數量增加。另一方面,陳舊的病灶則由密集增厚的纖維膠質斑塊組成,血管周圍偶爾可見淋巴和巨噬細胞;這時軸突仍相對保留完好。軸突的保留防止了沃勒變性。未受損的軸突可產生部分髓鞘再生,這是造成影像學上脫髓鞘斑「陰影修補」的組織學原因。在整個臨床過程中可見形狀、大小以及組織學改變新舊不同的各級病灶。
臨床表現
中樞神經系統散在分布的多數病灶與病程中呈現的緩解復發,症狀和體徵的空間多發性和病程的時間多發性構成了MS的主要臨床特點。多數病人呈急性或亞急性起病,少數起病緩慢。急性發病者較慢性發病預後好,我國MS患者急性或亞急性起病較多,MS臨床表現複雜。主要症狀有視力障礙、運動和感覺障礙或顱神經受累等。首發症狀以單眼或雙眼視力減退、肢體疼痛、感覺異常、奇癢及無力為最多見。痙攣性肢體癱瘓、小腦性共濟失調、傳導束性感覺障礙和膀胱功能障礙等。脊髓損害以後柱及側柱為常見,當累及頸段脊髓後柱時,患者屈頸時,在四肢或軀幹出現刺痛或過電樣感覺,稱為Lhermitte征,對診斷有一定價值,但並非特異性。
1 首發症狀
首發症狀包括一個或多個肢體局部無力麻木、刺痛感或單肢不穩,單眼突發視力喪失或視物模糊(視神經炎),復視,平衡障礙,膀胱功能障礙(尿急或尿流不暢)等,某些病人表現急性或逐漸進展的痙攣性輕截癱和感覺缺失。這些症狀通常持續時間短暫,數天或數周后消失,但仔細檢查仍可發現一些殘留體徵。眼部首發視力下降者約占30%~43%,病程中出現視力障礙者為68.6%~73%,視神經受累占70%~84%。病變多位於球後段視神經,其次為視交叉,故臨床上單眼視力首先受累者較多,呈進行性視力減退或突然視力下降至無光感。常見一過性復視,其特點是持續時間短、數小時或數日即可消失。一般在1~2天內視力下降,一周後即可恢復,6~8周後部分或全部恢復。眼底早期無異常,或視盤有輕度腫脹,隨後視盤顳側蒼白或全萎縮,視盤蒼白程度與視力減退不一定成比例。視野檢查有中心、旁中心或啞鈴狀暗點等。由於視交叉、視束和視放射的受累可引起同側或顳側偏盲,但少見。因病灶呈多發性,視野改變可極不規則。色覺障礙以紅綠色覺明顯,色相排列檢查法更易發現其異常。有紅綠色異常者,圖形VEP潛伏期延長更嚴重,且恢復較慢。MS的VEP潛伏期延遲與神經纖維的減少或軸突的變性及有髓神經纖維脫髓鞘病等因素有關,診斷上觀察圖形VEP潛伏期變化較觀察振幅改變敏感。圖形VEP潛伏期延遲與患者視力、視野及對比敏感度等視功能之間存在相關性。圖形VEP檢測有助於發現MS的隱匿型。
2 緩解期
首次發病後可有數月或數年的緩解期,可再出現新的症狀或原有症狀再發。感染可引起復發,女性分娩後3個月左右更易復發,體溫升高能使穩定的病情暫時惡化。復發次數可多達10餘次或更多,多次復發及不完全緩解後病人的無力、僵硬、感覺障礙、肢體不穩、視覺損害和尿失禁等可愈來愈重。
3 臨床常見體徵
多發性硬化患者的體徵多於症狀是重要的臨床特徵,患者主訴一側下肢無力、步態不穩和麻木感,檢查時卻可能發現雙側錐體束征或Babinski征。眼球震顫與核間性眼肌麻痹並存指示為腦幹病灶,是高度提示MS的兩個體徵。核間性眼肌麻痹患者病變眼內收不能,有分離性水平性眼球震顫,向外側注視時出現單眼水平性眼球震顫更明顯,而集合正常,乃內側縱束損害引起。腦幹的脫髓鞘斑塊也可引起其他顱神經功能障礙,以外展神經受累較多見,其次為動眼神經,滑車神經往往不受累。動眼神經障礙多呈不完全性,常見瞼下垂和瞳孔大小不等,不一定伴有眼球運動障礙。如有眼球運動障礙,則以向上、向下受限多見,說明病灶在動眼神經核或神經纖維尚未離開腦幹之前。其他的眼球震顫常由於腦幹或小腦病變引起,小腦病變時呈水平性或旋轉性眼球震顫,向病變側運動時更明顯。中腦病變時則可見垂直性注視癱瘓,向上注視時更明顯,其他還可見擺動性痙攣性眼球震顫。如中腦導水管、四疊體病變時可見Argyll-Robertson瞳孔。在運動後或體溫升高時發生視力下降,稱為Uhthoff征。近年來通過觀察,發現有時早期即有視盤周圍神經纖維層缺損,提示視網膜神經節細胞軸突潛在的萎縮,甚至比視力、色覺和視盤變化還早。對單側視力障礙嚴重或雙眼視力不對稱者、有相對性瞳孔傳入障礙、瞳孔周期時間延長等均有助於早期診斷視神經病變。
(1)肢體癱瘓多見,常見不對稱性痙攣性輕截癱,表現下肢無力或沉重感。
(2)約半數病例可見視力障礙,自一側開始,隔一段時間再侵犯另一側,或短時間內兩眼先後受累。發病較急,常有多次緩解-復發,可於數周后開始恢復。
(3)眼球震顫多為水平性或水平加旋轉,復視約占1/3。病變侵犯內側縱束引起核間性眼肌麻痹,侵犯腦橋旁正中網狀結構(PPRF)導致一個半綜合徵;其他腦神經受累少見,如中樞性或周圍性面癱,耳聾、耳鳴、眩暈、咬肌力弱、構音障礙和吞咽困難等。
(4)半數以上患者出現感覺障礙,包括深感覺障礙和Romberg征。
(5)約半數病例可見共濟失調,但Charcot三主征(眼震、意向震顫和吟詩樣語言)僅見於部分晚期MS患者。
(6)神經電生理檢查證實,MS可合併周圍神經損害(如多發性神經病、多發性單神經病),可能因周圍神經P1蛋白與中樞神經系統的MBP為同一組分,均發生脫髓鞘所致。
(7)可出現病理性情緒高漲如欣快和興奮,多數病例表現抑鬱、易怒,也可見淡漠、嗜睡、強哭強笑、反應遲鈍、重複語言、猜疑和迫害妄想等精神障礙。 晚期病例檢查時常發現視神經萎縮、眼球震顫和構音障礙、某些或全部肢體可出現錐體束征、感覺或小腦體徵。已經確認某些症狀在MS極為罕見,如失語症、偏盲、錐體外系運動障礙、嚴重肌萎縮和肌束顫動等,常可作為MS的除外標準。
4 發作性症狀
除上述神經缺失症狀外,MS的發作性症狀也不容忽視。例如,Lhermitte征是過度前屈頸部時出現異常針刺樣疼痛,自頸部沿脊柱放散至大腿或足部,是頸髓受累徵象。球後視神經炎和橫貫性脊髓炎通常可視為MS發作時的表現,也常見單肢痛性痙攣發作、眼前閃光、強直性發作、陣發性瘙癢、廣泛面肌抽搐、構音障礙和共濟失調等。但這些極少以首發症狀出現,傾向以固定模式在數天、數周或更長時間內頻繁再發,可完全緩解。某些以罕見症狀或非常規方式起病的MS病例常使診斷困難,如年輕患者典型三叉神經痛,特別是雙側性應高度懷疑MS。
5 視神經脊髓炎和橫斷性脊髓炎
視神經脊髓炎和橫斷性脊髓炎兩個特殊綜合徵,是MS最典型的發病模式,也是建立MS診斷的特異性依據
(1)視神經炎
約有25%的MS患者(在兒童比例更大)球後或視神經炎是首發症狀。其特點為急性發展,在數小時或數天內單眼部分或全部失明。部分患者在視力喪失前1~2天有眶周疼痛,疼痛可因眼球運動或觸壓眼球而加劇。少數患者視力減退在數月內進行性發展,類似壓迫性病變或視神經固有腫瘤的表現。常發現黃斑區暗點和盲點(偏心)。也常見其他範圍不同的視野缺陷,甚至可為偏盲、同象限性盲。有些病例同時或幾天或幾周內雙側視神經受累。有1/8的患者將重發。約有一半患者有視盤腫脹、水腫(視盤炎)。視盤炎出現與否取決於脫髓鞘病損距視盤的距離。視盤炎和因顱內壓增高所致的視盤水腫不同,前者常表現為嚴重而突發的視力喪失。視神經事實上是大腦傳導束的一部分。視神經受累符合MS僅侵犯CNS的原則。
約1/3的視神經炎患者完全恢復,剩餘的大部分即使有嚴重的視力減退和視神經盤蒼白也可有明顯的改善。色覺障礙常持續存在。視力改善一般在發病後2周,或在經皮質激素治療後不久。一旦神經功能開始改善,在數月內可持續好轉。
1/2或更多的單純視神經炎患者最終發生MS其他的症狀和體徵。如果首次視神經炎發作於兒童期,發展為MS的危險性最低(提示一些在兒童期發病的疾病類型不同)。Rizzo和Lessel在一項前瞻性調查中發現,74%女性患者和34%的男性患者在視力喪失發病15年後發展為MS。觀察時間越長、檢查越細緻發現最終發展成為MS的比例越高。多數在首次發作的5年內出現其他症狀。實際上,很多臨床為單純視神經炎的患者,MRI發現大腦白質有MS病灶,說明無症狀播散性病損已存在。
常見的視神經炎的病理基礎是脫髓鞘性改變。血管炎損傷或由於腫瘤、囊蟲對視神經的壓迫很少引起中心或偏心盲點。
(2)急性橫貫性脊髓炎
是常見的一種脊髓受累的急性炎性脫髓鞘病變,無論是單一急性病程還是慢性(多發)病程類型,在多數情況下被視為MS的一種表現形式。
該病的臨床特點是快速出現下肢癱瘓、軀幹感覺平面、括約肌功能障礙和錐體束征。CSF呈中度淋巴細胞增高和蛋白升高,但在疾病的初期階段腦脊液可為正常。1/3的患者在發病前數周內有感染性疾病史,這種情況多為感染後所致單相脫髓鞘性病變。不到一半的患者在脊髓發病的同時有其他神經系統無症狀性病灶,或者5年內發現彌散的臨床症狀。因此,急性橫貫性脊髓炎較視神經炎與MS相關性為小。
同一部位復發性脊髓炎,經細緻的MRI檢查未發現有其他部位脫髓鞘病灶的患者引起人們的注意。部分病例甚至在腦脊液中出現寡克隆帶。此種情況臨床不少見。多數人同意,這是一種局限性、復發性的脊髓型MS。值得一提的是,單純的復發性脊髓炎偶爾伴有紅斑性狼瘡,合併有結締組織病、抗磷脂抗抗體綜合抗體綜合徵或有其他自身抗體的存在。同樣,視神經炎也有僅局限於視神經的多次復發。
一旦MS的診斷成立,可發現數個臨床綜合徵規律地出現。約1/2的患者為混合或全身型,臨床表現為視神經、腦幹、小腦和脊髓受損的症狀和體徵;另30%~40%的患者顯示為不同程度痙攣性共濟失調和四肢末端深感覺障礙,基本符合脊髓型MS。非對稱性痙攣性下肢癱瘓是進展型MS最常見的表現形式。小腦或腦橋延髓小腦型和全盲型各占5%。因此,混合型和脊髓型約占臨床病例的80%。
MS患者常表現有精神異常,部分病例表現為欣快。更多的病例表現為抑鬱、易激惹和脾氣暴躁。其他精神錯亂如保留記憶力喪失、全面性痴呆或精神混亂狀態可有一定規律地發生於疾病的後期。MS的認知障礙較符合前面所述的「皮質下痴呆」。有嚴重意志缺失的額葉綜合徵是晚期MS常見特徵。2%~3%的MS患者在其病程的某一時期有一次或反覆的癇性發作,這是由大腦皮質或臨近皮質的病症引起。
6 其他變異型MS
(1)急性多發性硬化
急性多發性硬化是罕見的惡性型MS,表現為在幾周內大腦、腦幹、脊髓聯合受累,導致患者呈木僵、昏迷或去大腦狀態,有明顯腦神經和皮質脊髓束異常,症狀進行性發展可在幾周或數月內死亡。屍解發現肉眼可見的典型急性MS斑。惟一與一般型MS不同的是多數硬化斑的新舊一致,許多靜脈周圍髓鞘脫失區融合明顯。通常CSF細胞反應活躍(細胞數增加)。
(2)多發性硬化並發周圍神經病
MS的患者可同時伴有多發性周圍神經病或各種各樣的單神經病。這種關係帶來一些推測和矛盾。這種結合的偶發性提示MS和周圍神經病發生的可能是巧合,但又難以解釋後者為什麼是一種非常具有特徵的周圍神經病。中樞和周圍神經都可發生自身免疫性脫髓鞘改變,發生在後者導致慢性、炎性多發性周圍神經病。當然,根性和周圍神經性運動和(或)感覺症狀也可因累及脊髓神經根進入區或脊髓腹側白質內穿出纖維。在MS的後期有患維生素缺乏性周圍神經病的可能。[2][3]
輔助檢查
1、 腦脊液常規
約1/3的MS患者,特別是急性發病、惡化型的病例,腦脊液常有輕度到中度的單核細胞增多(通常少於50×106/L)。在進展型視神經脊髓炎患者和某些腦幹脫髓鞘疾患,細胞總數可達100×106/L。在急性高峰病情下,細胞比例可以多核白細胞為主。細胞數增減反映疾病的活動性。
約40%的患者,CSF中的總蛋白含量升高。蛋白增多是輕度的,蛋白的濃度超過100mg/dl在MS很少見。更為重要的是在2/3的患者γ-球蛋白(主要為IgG)的比例增加(超過總蛋白的12%)。
2、 IgG指數
另一種診斷是通過測定血清和腦脊液中白蛋白和γ-球蛋白的比值監測IgG指數。比值大於1.7為可能性MS。已經證明MS患者CSF中γ-球蛋白是在CNS合成的,在瓊脂電泳中被分離有異常泳帶,稱為寡克隆帶(IgG)。這種寡克隆帶也可見於梅毒和亞急性硬化全腦炎患者的CSF。
CSF出現的寡克隆帶不會在血液中出現,這對早期診斷非典型MS具有特殊的意義。具有寡克隆帶的首次發作的MS可預測為慢性復發性MS。很多患者在急性期CSF中含有高濃度的髓鞘鹼性蛋白(MBP);在慢性進展期MBP含量較低或正常;在疾病的緩解期MBP含量正常。其他破壞髓鞘的病損(如梗死)也可增加MBP的水平。因此,這一測定指標對診斷無特異性。
將細胞數、總蛋白、γ-球蛋白和寡克隆帶都考慮在內,大多數患者會發現腦脊液異常。目前,測定CSF中作為總蛋白一個組分的γ-球蛋白和寡克隆帶是MS最可靠得化學檢測方法。
3、 電生理檢查
當臨床資料提示CNS僅有一個病灶時,數種生理性和放射性檢查可能顯示無症狀性病灶的存在,這種情況常見於疾病的早期或脊髓型MS。這些檢查包括視、聽和軀體感覺誘發反應;電眼圖;瞬目反射改變;視成像的閃光融合變化。據報道MS患者中50%~90%的患者有一項或多項以上檢查異常。視誘發的異常率在確診的MS患者中為80%,在可能或可疑MS中占60%;軀體誘發的異常率在兩組病例中分別為69%和51%;腦幹聽誘發異常率(一般波間潛伏期延長或波5峰值降低)分別為47%和20%。*[4]
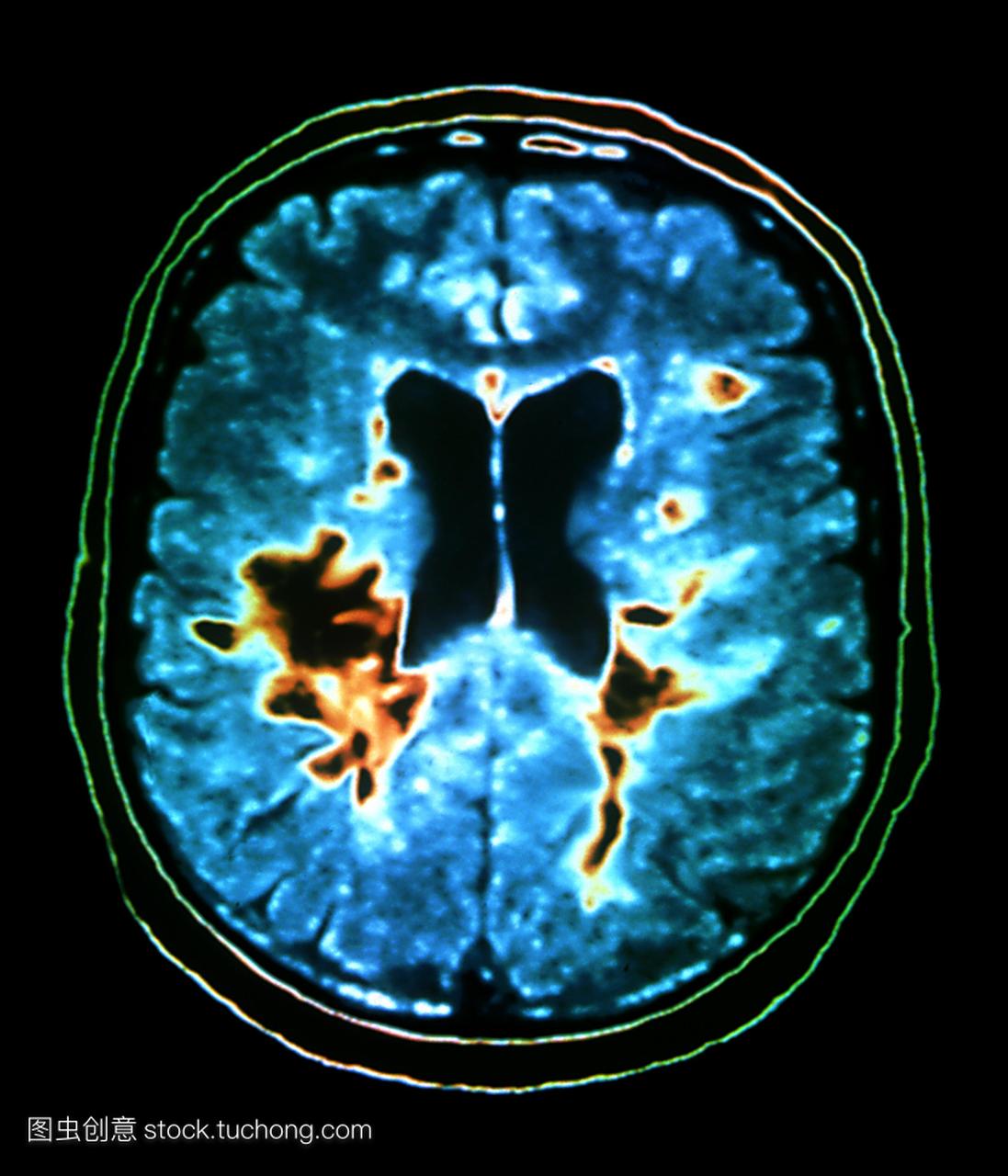
4、 影像學檢查
MRI比CT更為敏感地顯示大腦、腦幹、視神經和脊髓的無症狀性多發性硬化斑。80% MS病例有多發性病灶。應該指出腦室周圍信號增強可見於多種病理過程,甚至見於正常人,特別是老齡人。在後者,腦室周圍的改變較MS時程度較輕、邊界較平滑。散在的大腦多發性硬化病灶並無特異的MRI表現。在T2加權像可見數個非對稱性、邊界清楚、緊靠腦室表面的病灶,通常提示Ms。於纖維走行相應、放射狀分布的脫髓鞘性改變特別支持診斷。有些病灶在急性期可通過雙倍或三倍正常量的釓(gadolinium)顯示強化。連續的MRI檢查可顯示疾病的發展。[5]
診斷標準
1.病灶 臨床上有二個或二個以上中樞神經系統白質內好發部位的病灶如視神經、脊髓、腦幹等損害的客觀體徵;
2.病程 呈緩解和復發。二次發作間隔至少一個月,每次持續24小時以上,或階段性進展病程超過半年;
3.起病年齡 在10~50歲間;
4.其他原因的排除 如腦瘤、腦血管性疾病、頸椎病等。
四項標準均具備者則可診斷為「臨床確診」;如1、2缺少一項者,則診斷「臨床可能是多發性硬化」;如僅有一個好發部位首次發作,則只能作為「臨床可疑」。其他腦脊液中IgG指數增高以及IgG單克隆帶的出現、血清抗磷脂抗抗體陽性、肝病壞死因子活性增高、髓鞘鹼性蛋白增高等可作為參考。
鑑別診斷
MS常和下列疾病鑑別:
1、 播散性腦脊髓炎
播散性腦脊髓炎是一種廣泛散在性病損的急性疾病,具有自限性,多為單一病程。另外,該病常有發熱、木僵和昏迷,而這些特徵在MS很少見。
2、 系統性紅斑狼瘡和其他少見的自身免疫性疾病(混合性結締組織病,舍格倫綜合徵,硬皮征,原發性膽汁性肝硬化)
在CNS白質可有多個病灶。這些疾病的CNS損傷與潛在的免疫性疾病的活動性或諸如針對自身DNA或磷脂的自身抗體的水平相平行。多先有或合併有其他系統性損害,但也有脫髓鞘或大腦半球的病損先於其他系統器官的例子。5%~10%的MS患者攜帶抗核或抗雙鏈DNA抗體而沒有狼瘡或其他系統損害的表現。
3、 白塞病
白塞病以再發性虹膜睫狀體炎、腦膜炎、黏膜及生殖器潰瘍;關節、腎、肺部症狀以及大腦多發性病灶為鑑別特徵。
4、 脊髓壓迫征
單純的脊髓型MS常伴有不同程度的後索受累。進行性痙攣性輕截癱的單純脊髓型MS的診斷特別困難。前面已經提到,脊髓型MS特別傾向於影響老年女性,這種情況應仔細排除因腫瘤或頸椎關節病變而致的脊髓壓迫征。在病程某段時間出現根性疼痛常是脊髓壓迫征的表現,在MS則少見。頸部疼痛、活動受限和因神經根受累引起的嚴重肌肉萎縮可見於脊椎關節病變而MS很少有以上症狀。作為一般規律,在脫髓鞘性脊髓病的早期可見腹部反射消失,男性出現陽痿和膀胱功能障礙,而在頸椎關節增生的病例上述症狀則出現在晚期,或者根本不出現。CSF蛋白含量在脊髓壓迫征可顯著升高。但無其他MS特異性蛋白的異常。最有價值的鑑別方法是MRI和CT脊髓顯影術,任何神經系統體徵僅限於脊髓的進行性痙攣性輕截癱,都應進行脊髓顯影。
5、 顱底凹陷症和扁平顱底
顱底凹陷症和扁平顱底患者短頸,放射檢查可確定診斷。因顱底骨發育畸形以及顱骨孔、腦橋小腦角、斜坡和其他顱後窩腫瘤等引起的神經綜合徵也可被誤診為MS。在上述情況,一個孤立而部位特別的病灶可引起腦幹、小腦、後組腦神經和上頸髓的神經症狀和體徵,而易被認為是一播散性病損。如患者所有的症狀和體徵可以用神經軸某一區域的一個病灶解釋,就不應診斷MS,是臨床應遵循的規則。
6、 遺傳性共濟失調
偶爾MS可與遺傳性共濟失調相混淆。後者常有家族史及其相關的遺傳特性,呈隱匿性發病、慢性持續進展,具有對稱性和特異臨床方式。腹壁反射和括約肌功能完好、弓型足、脊柱後側凸、心臟病是支持遺傳性疾病的一些常見特徵。
治療
近年來的治療實驗大多基於抗炎和免疫抑制藥物。臨床對照研究證明,只有促皮質素(促腎上腺皮質激素)、甲潑尼龍(甲基強的松龍)、潑尼松(強的松)、環磷酰胺和干擾素(β-干擾素)對改善臨床和MRI病損作用良好。在抗炎因子的干預下,患者從每次發作中恢復的速度加快。但在急性惡性型MS,大部分患者抗炎治療是無效的;少數患者療效僅能維持1個月余。尚不能證明類固醇激素能縮短整個病程,或者能夠預防復發,所以對其長期的療效難以定論。
1、 皮質激素
關於皮質激素的應用劑量,首次大劑量是至關重要的。靜脈給予大劑量甲潑尼龍(甲基強的松龍)(500mg/d,3~5天),後口服較大劑量潑尼松(強的松)能有效的緩和急性或亞急性MS以及視神經炎,能夠縮短其病程。如不能靜脈用甲潑尼龍(甲基強的松龍),可用口服潑尼松(強的松)代替,從60~80mg/d開始,這樣可避免住院治療。對於嚴重的發作,特別是脊髓炎對大劑量靜脈用藥反應更快。
在急性視神經炎的治療中避免用口服。提倡將皮質激素的治療限制在3周內,如果症狀反覆,延長減量過程。這種短期皮質激素治療副作用較少,但仍有部分患者可出現失眠,個別可出現抑鬱或躁狂症狀。療程達數周以上的患者,易出現高血壓、高血糖和糖尿病失控、骨質疏鬆、髖關節無菌性壞死、白內障和少見的消化道出血、活動性結核。適當補鉀是必要的。
2、 免疫調節藥
曾試用過多種免疫調節藥,僅少數藥物如硫唑嘌呤和環磷酰胺有效,另外對一小部分患者給予全身淋巴放射治療似乎能改善部分病情。這些治療方法能改善臨床症狀支持MS的CNS損傷的機制是自身免疫過程的學說。然而,長期免疫抑制藥應用的危險性,如癌變大大限制了這類藥物的廣泛應用。
有兩種新的治療方法有希望改變MS的自然病程。初期臨床試驗表明,皮下注射干擾素(β-干擾素)能降低MS復發的頻率和嚴重程度,減少了MS病灶數目。有證據說明干擾素(β-干擾素)能降低大腦半球脫髓鞘改變。該藥能否阻遏神經功能障礙的進展有待進一步驗證。然而,臨床療效並不令人振奮。Bornstein等也報道了MBP多聚體和複合多聚體I(Cop I)對復發緩解型MS有效。這種藥物尚待FAD批准。對新近通過口服牛髓鞘使髓鞘脫敏的試驗尚未得出結論。
3 神經營養藥物
胞二磷膽鹼(250mg肌注1次/d)鹼性成纖維細胞生長因子(DFGF1600u 肌注1次/d)可酌情選用。
4 對症治療
對痛性強直發作、三叉神經痛、癲癇發作者可用卡馬西平0.13次/d,痙攣者可給安定等。
5、 一般治療
一般措施包括保證適當的臥床休息時間、避免過度疲勞和減少感染,爭取從首發或病情惡化中最大程度的恢復。利用可能的康復措施(如拉帶、輪椅、滑道、電梯等)儘量拖後疾病的臥床時間。精心護理、利用變換壓力床墊、硅膠墊和其他特殊設備預防臥床期褥瘡的發生。疲勞是MS患者常見的主訴,特別在急性發作期,金剛烷胺(100mg早、晚各一次)或匹莫林(pemoline)(晨一次口服20~75mg)可緩解疲勞症狀。
膀胱功能障礙是治療中較難以處理的問題。其中主要的症狀是尿瀦留,氯貝膽鹼(比賽可靈)對此可能有幫助。在尿瀦留時,為避免感染應監測殘餘尿量,殘餘尿量不能超過100ml。另一個常見的問題是尿急、尿頻(膀胱痙攣)。溴丙胺太林(溴化西胺太林,普魯苯辛)或奧昔布寧(尿多靈,氯化羥丁寧,Ditropan)能鬆弛逼尿肌可緩解這一症狀,這類藥物最好間斷應用。間歇性導尿對具有嚴重膀胱功能障礙,特別是尿瀦留者是非常必要的。患者可學會自己導尿,從而減少保留尿管所帶來的感染危險。嚴重便秘時,最好進行灌腸。直腸規律性訓練對保持大便通暢有幫助。
對嚴重的痙攣性截癱和下肢痛性屈曲痙攣患者,以及其他一些痙攣狀態,通過置留管或埋藏泵鞘內注射巴氯芬(巴氯酚)有一定的療效。輕度痙攣者可口服baclofen。以上方法無效時,背部脊神經根切斷術、脊髓切斷術、閉孔神經壓榨術等外科方法可使症狀長期緩解。
對下肢輕微運動誘發的非常嚴重、致殘性震顫可行丘腦腹外側切除術。卡馬西平、氯硝西泮(氯硝安定)對此症狀也有一定的作用。[6]
視頻
圖說多發性硬化
2分鐘神經科學 - 多發性硬化症
多發性硬化康復指導
參考資料
- ↑ 王維治. 多發性硬化的研究進展及現狀(述評). 中國神經免疫學和神經病學雜誌. 2001, (03): 3-5+10.
- ↑ (美)貝沙. 多發性硬化手冊. 遼寧科學技術出版社. 2003年. ISBN 9787538139105.
- ↑ 黃德暉, 吳衛平, 蒲傳強; 等. 多發性硬化226例臨床分析. 中國神經免疫學和神經病學雜誌. 2003, (03): 14-17.
- ↑ 吳宗忠 陳躍鴻. 多發性硬化的臨床與視覺誘發電位. 2005-9-22 [11 三月 2020] (中文).
- ↑ 王飛; 於春水; 李坤成. 多發性硬化MRI研究進展. 中國醫學影像技術. 2009, (11).
- ↑ 郭怡菁; 胡學強. 多發性硬化臨床免疫干預治療研究現狀(綜述). 中國神經免疫學和神經病學雜誌. 2001, 8 (4): 228-230.